Controlling the Speed of Rowan's Docking
by Corin Wagen · Sept 22, 2025

View of the Entrance to the Venetian Arsenal, by Canaletto (1732)
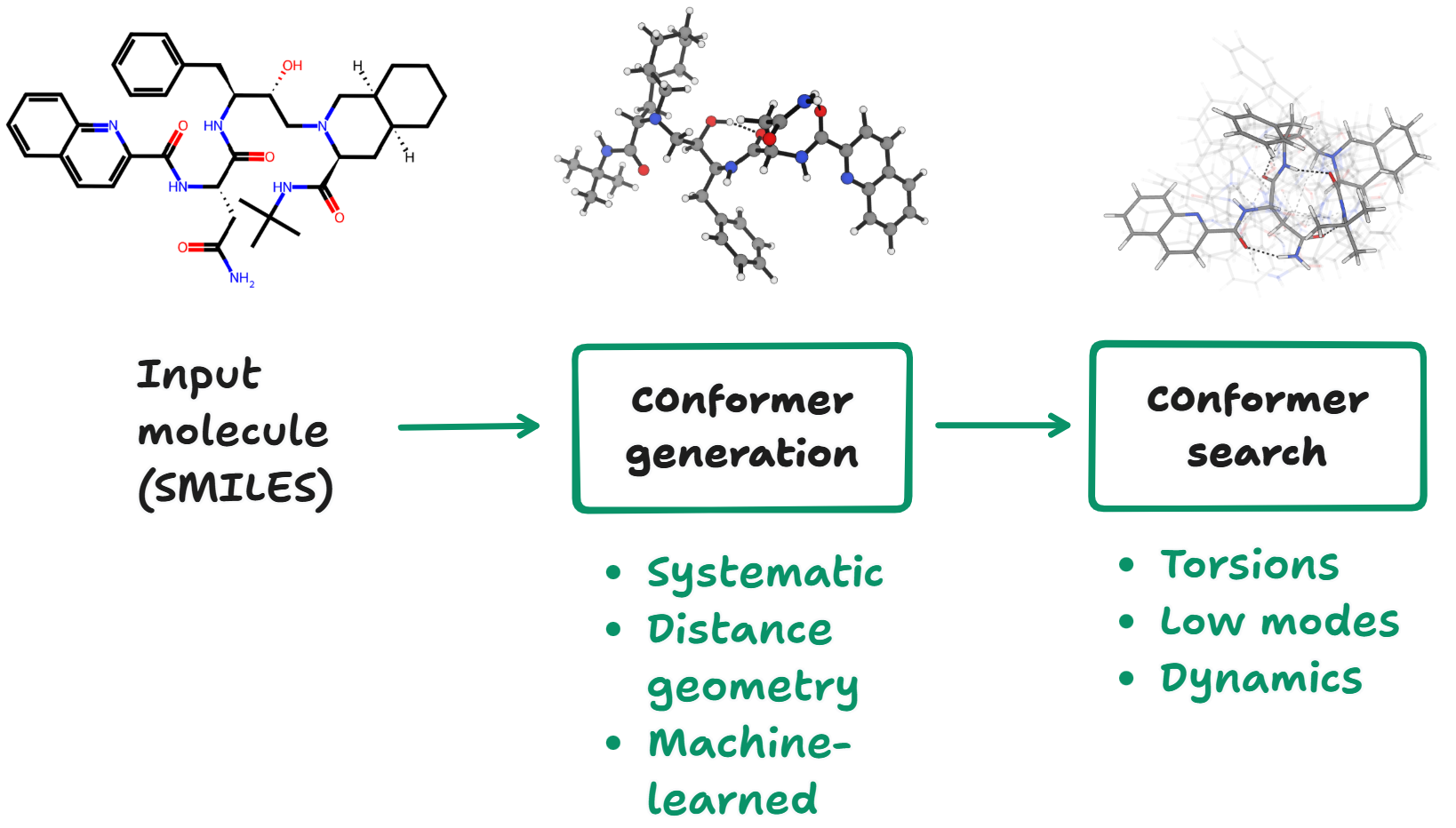
Rowan uses AutoDock Vina to run docking (with an exhaustiveness of 8 by default). Through Rowan's Python API there are three readily accessible options that can be selected to tune how docking is run:
do_csearch, which controls whether a conformer search is run before docking (using ETKDG)do_optimization, which controls whether the input conformers are optimized before docking (using AIMNet2)do_pose_refinement, which controls whether the output poses are optimized (again using AIMNet2)
By default, docking calculations run through the web interface have all of these options enabled, which makes docking calculations more thorough but slower. This is ideal for cases in late-stage optimization when a thorough assessment of a given compound is desired, but makes Rowan's docking too slow for many high-throughput screening contexts; for virtual screens it often makes sense to disable some or all of these steps.
To illustrate the effects that each of these settings can have, we ran a bunch of docking calculations. We selected PDE4 as our protein target (based on a sanitized 1XM6 structure) and docked the known PDE4 inhibitor rolipram against it.
Here's the script we used, which takes a local sanitized PDB file with a known pocket and iteratively tries different combination of settings:
from pathlib import Path
import rowan
from rowan.protein import upload_protein
import stjames
rowan.api_key = "rowan-sk-your-secret-key-here"
protein = upload_protein("PDE4 cleaned from 1XM6", Path("pde4.pdb"))
pocket_specification = [[46.37, 1.64, -9.88], [19.2, 13.37, 17.72]]
ligand_smiles = "COc1ccc([C@@H]2CNC(=O)C2)cc1OC1CCCC1"
ligand = stjames.Molecule.from_smiles(ligand_smiles)
def run_docking(
do_optimization: bool,
do_conformer_search: bool,
do_pose_refinement: bool,
) -> tuple[int, int, float, float]:
"""
Run a docking calculation through Rowan and return various metrics.
"""
workflow = rowan.submit_docking_workflow(
protein,
pocket_specification,
initial_molecule=ligand,
do_csearch=do_conformer_search,
do_optimization=do_optimization,
do_pose_refinement=do_pose_refinement,
)
workflow.wait_for_result()
workflow.fetch_latest(in_place=True)
num_conformers = len(workflow.data["conformers"])
num_poses = len(workflow.data["scores"])
best_score = min(s["score"] for s in workflow.data["scores"])
total_time_s = workflow.elapsed
return num_conformers, num_poses, best_score, total_time_s
for do_optimization in [True, False]:
for do_conformer_search in [True, False]:
for do_pose_refinement in [True, False]:
num_confs, num_poses, best_score, time_s = run_docking(
do_optimization,
do_conformer_search,
do_pose_refinement
)
print("Docking run finished:")
print(f"\tOptimization? {do_optimization}")
print(f"\tConformer search? {do_conformer_search}")
print(f"\tPose refinement? {do_pose_refinement}")
print(f"\t{num_confs} conformers, {num_poses} output poses")
print(f"\tBest score: {best_score:.3f}")
print(f"\tTotal elapsed time: {time_s:.1f} seconds")
Running the above script gave a clear comparison of each docking method's performance, which we organized into the below table. (We excluded rows where do_csearch is set to True and do_optimization is False because optimization is automatically run in these cases—a message is printed in the logfile when this happens.) There are obviously slight run-to-run variations in timing and outputs, but we've found that the values shown below are pretty consistent, and that timings generally only vary by a second or two per run.
| # | Optimization? | Conformer search? | Pose refinement? | Conformers | Poses | Best Score | Time (s) |
|---|---|---|---|---|---|---|---|
| 1 | Yes | Yes | Yes | 5 | 20 | -6.842 | 194.1 |
| 2 | Yes | Yes | No | 5 | 19 | -6.828 | 112.6 |
| 3 | Yes | No | Yes | 1 | 4 | -6.630 | 29.7 |
| 4 | Yes | No | No | 1 | 4 | -6.630 | 20.2 |
| 5 | No | No | Yes | 1 | 4 | -6.727 | 29.3 |
| 6 | No | No | No | 1 | 4 | -6.727 | 17.6 |
Looking at the above data, a few conclusions are apparent:
- Running a conformer search before docking allows Rowan to find a slightly better pose—albiet at the cost of a significantly slower workflow. Rows where conformer searches were run (1 & 2) all found a lower docking score but took roughly 4–6x as long as the rows without conformer searches.
- Running optimizations before docking is very fast—row 3 and row 5 take virtually the same amount of time, despite row 3 having optimization set to
Trueand row 5 having optimization set toFalse. Interestingly, optimizing the ligand's structure leads to a worse docking score here. - Pose refinement adds a non-trivial amount of time to the overall workflow runtime: a few seconds per pose for this system. While pose refinement matters a lot if the actual ligand conformations are going to be visually inspected (or if strain calculations are going to be run), it should probably be omitted for high-throughput virtual screening.
The fastest possible settings (row 6) allow a full protein–ligand docking calculation to be run in under 20 seconds, which is quick enough for most high-throughput screening applications.