Testing Rowan on the OpenBind EV-A71 Release
by Corin Wagen · May 6, 2026
The UK-backed OpenBind project recently released their first public batch of data, focusing on the Enterovirus A71 (EV-A71) 2A protease. OpenBind released, in their words, "a structure–affinity dataset containing 925 crystallographic binding events from 699 compounds, and associated affinity measurements for 601 compounds." All data is permissively licensed and publicly available as of today.
Here's the OpenBind team's explanation of what makes this dataset different (emphasis added):
This [dataset] is not just a collection of isolated protein–ligand complexes. It links many related compounds, crystallographic binding modes, and affinity measurements. This makes the dataset useful for training, fine-tuning, benchmarking, and error analysis: users can ask whether predicted poses match observed binding modes; whether related compounds preserve, shift, or lose interactions; whether affinity models capture local structure–activity relationships; how docking and cofolding methods behave across a consistent experimental series; and whether fragment-screen data can improve prediction on follow-on compounds.
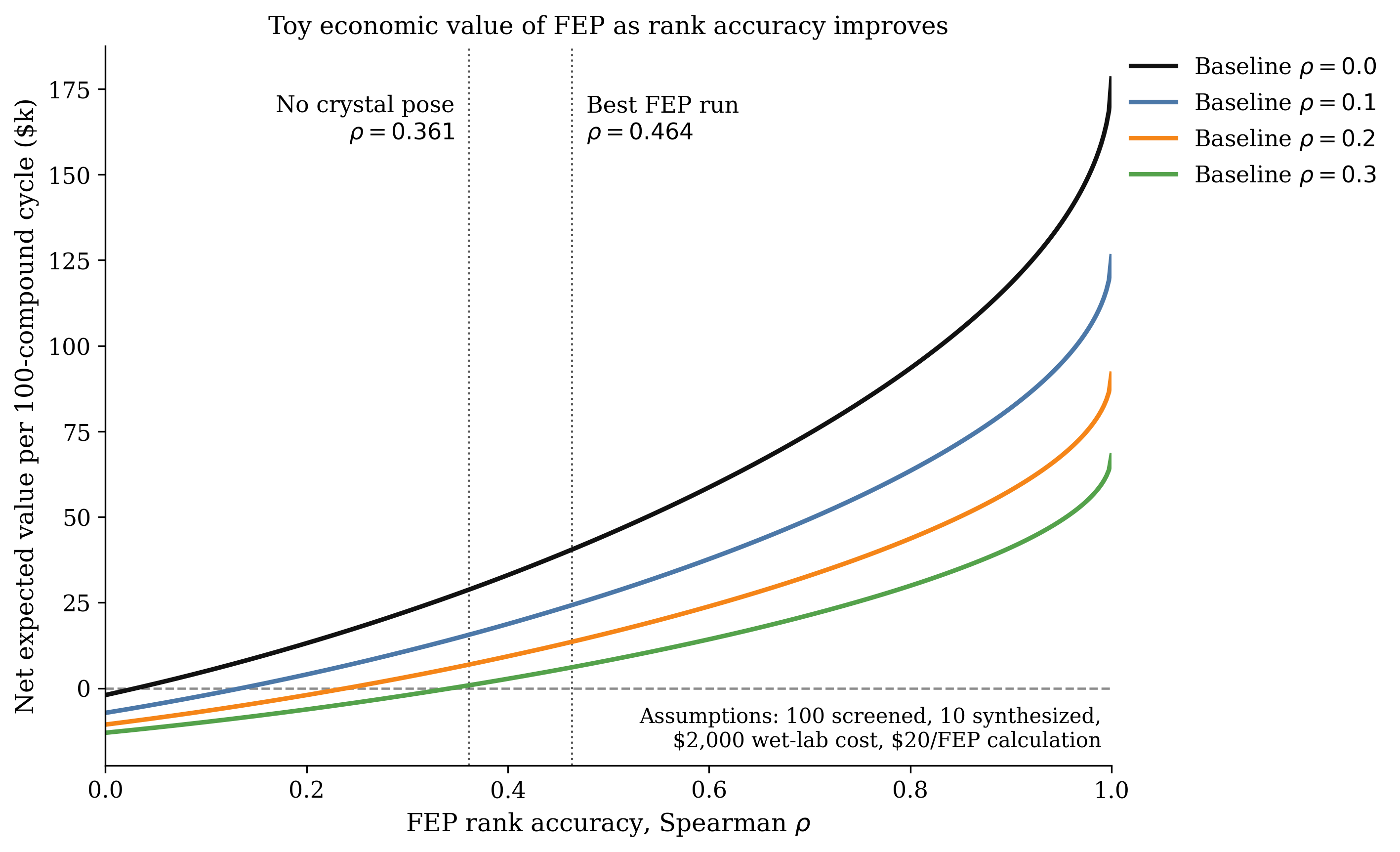
We wanted to assess how well Rowan's FEP pipeline fared against this dataset; although the full set of compounds was too diverse to test, we picked a subset of 76 molecules with a shared thiopyrimidine core to serve as a benchmark subset. This subset was particularly well-suited for FEP: all compounds bound in the same pocket, there's a substantial dynamic range (almost 4 kcal/mol), and the protein conformation was very similar between all bound poses (~0.2 Å RMSD for alpha carbons), which eliminates potential error from target structural reorganization. (The full dataset used is available below in the Rowan workflows.)
Pose Retrieval
The OpenBind dataset contains binding affinities and crystal structures for every single compound. This is an uncommon situation—typically, only a small number of compounds have experimental crystal structures, and these experimental structures are used to inform docking protocols for the remaining compounds. We envisioned that we could test our analogue-docking workflow by generating poses in silico and seeing how close Rowan's predicted poses were to the experimentally determined structures.
We started from an arbitrary compound (x7259a) and docked the rest of the compounds against this (starting only from a SMILES representation of these compounds). Like most docking workflows, Rowan's analogue-docking workflow returns an ensemble of poses for each structure. For prospective cases like FEP where we have to pick a single pose to continue forward with, we typically pick the pose with the lowest docking score (unless a scientist manually overrides this). We thus scored accuracy two ways: (1) how close is the top-ranked pose predicted by Rowan to the crystallographic structure, and (2) how close is the closest pose in the ensemble to the crystallographic structure?
Here's how we did:
| Analysis | ≤ 0.5 Å | ≤ 1.0 Å | ≤ 2.0 Å | ≤ 3.0 Å |
|---|---|---|---|---|
| Single pose | 21/76, 27.6% | 40/76, 52.6% | 69/76, 90.8% | 72/76, 94.7% |
| Ensemble | 44/76, 57.9% | 68/76, 89.5% | 75/76, 98.7% | 76/76, 100.0% |
We found that Rowan generated poses within 1.0 Å of the crystallographic structure over 50% of the time, even when we only chose a single pose. When we compared to the top-ranked pose in the ensemble, our success at 1.0 Å increased to almost 90%. Loosening cutoffs increased success rates; 91% of single poses and 99% of the ensembles matched at a 2.0 Å cutoff (the typical threshold for e.g. docking or co-folding algorithms).
These data are encouraging. Although we're not able to perfectly retrieve experimental structures, as demonstrated by the poor success rates at 0.5 Å, we're reliably able to generate structures for most molecules that are good enough for downstream use cases.
Free-Energy Perturbation
Since OpenBind has both crystal structures and affinity values, we envisioned comparing the Rowan-generated poses to the crystallographic poses as inputs to RBFE calculations. Accordingly, we ran free-energy perturbation starting from both the crystallographic poses and the top-ranked poses predicted by Rowan. (These are the best poses by docking score, not necessarily the poses which are closest to the crystal structure, again to mimic a scenario where we wouldn't have crystal structures for every single ligand.)
We were unsure a priori what to expect from this experiment. Since Rowan's analogue-docking workflow enforces rigorous maximum-common-substructure alignment, the Rowan-docked poses were more tightly superimposed than the crystallographic poses (see below image), which might lead to better converged FEP calculations. On the other hand, the crystallographic poses are likely more representative of the experimental bound-state conformations, and more physical poses ought to lead to better-quality binding-affinity values. As we'll see in a second, the performance on both sets was quite poor, making it difficult to compare the performance of crystallographic and docked poses.
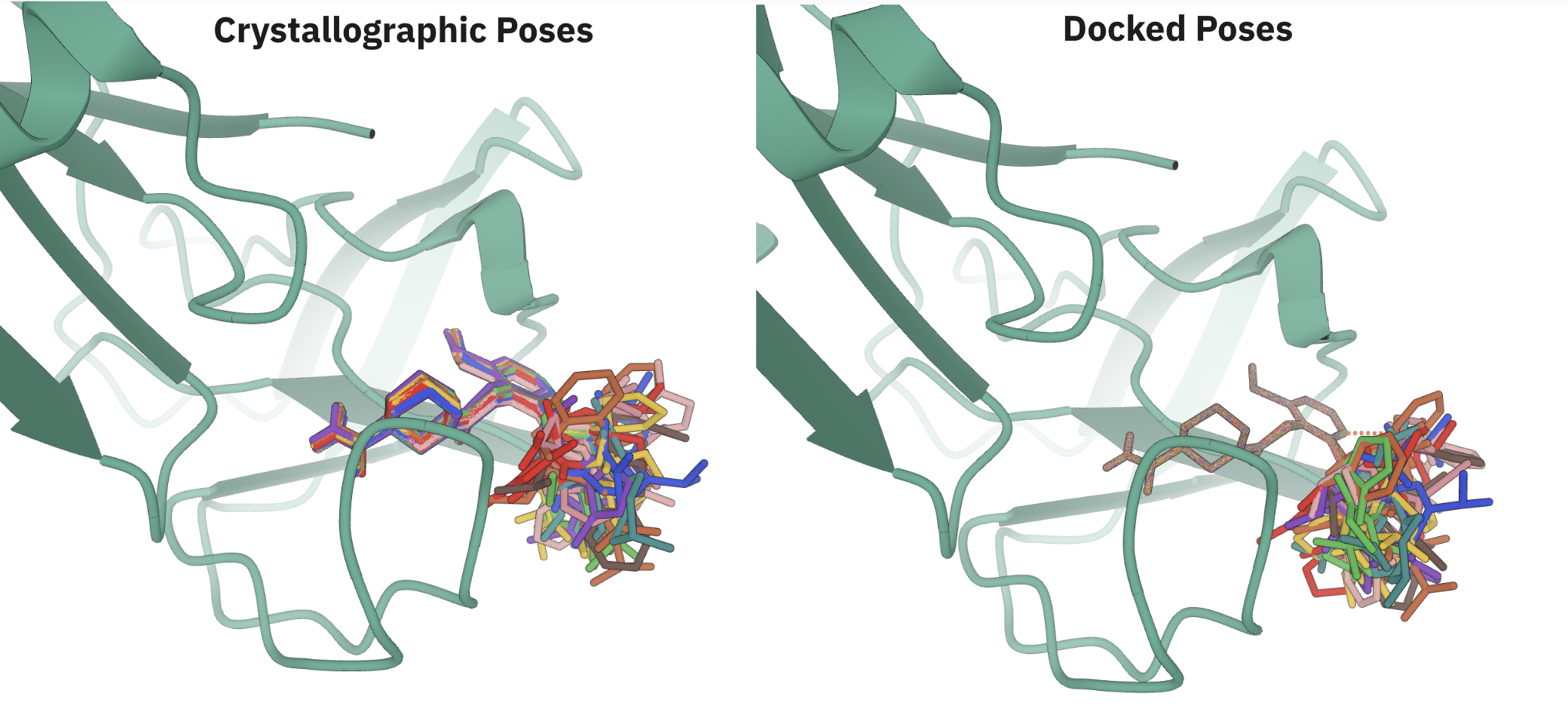
Visual comparison of crystallographic and Rowan-predicted poses.
Both FEP runs took about 12 hours on Rowan's standard 4 L40S GPU hardware, which amounts to about 5–6 minutes per leg (or 10 minutes per ligand). We found that the FEP run starting from crystal structures performed fine (link to Rowan workflow), with an MAE of about 1 kcal/mol but poor ranking-based accuracy:
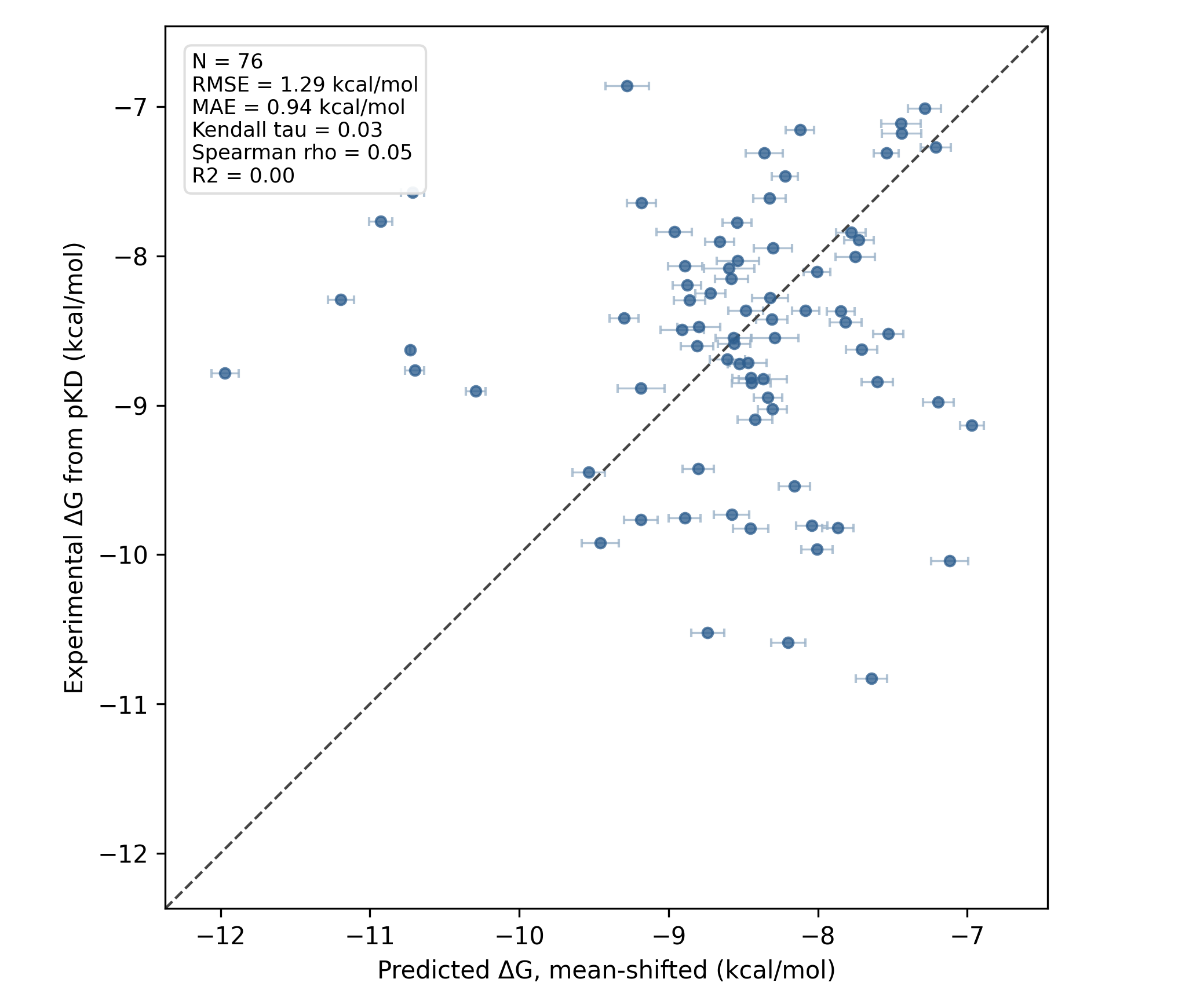
RBFE results starting from crystallographic poses.
The FEP run starting from Rowan's analogue-docking workflow did slightly "better" (in terms of error), with an MAE of 0.8 kcal/mol (link to Rowan workflow):
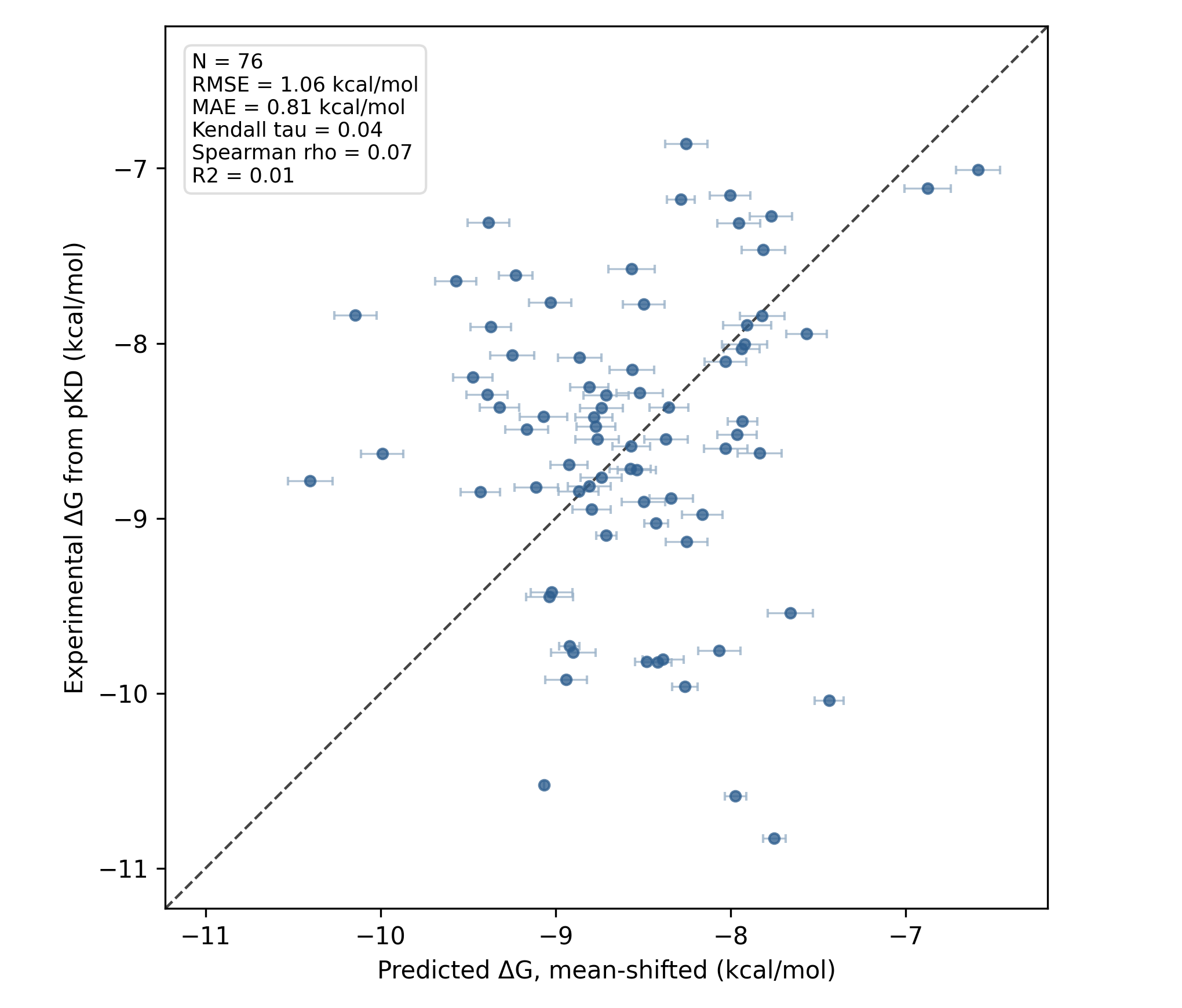
RBFE results starting from Rowan-generated poses.
While both runs performed decently in terms of error (in line with established FEP benchmarks), neither run gives satisfactory ranking-based accuracy. This is unfortunately a pretty common story in real-world FEP deployments; while many procedures seem good on reported public benchmarks, prospective FEP usage can struggle at first and often takes a number of iterations to get right.
| Input poses | N | MAE (kcal/mol) | RMSE (kcal/mol) | Spearman ρ | Kendall τ | R² |
|---|---|---|---|---|---|---|
| Crystallographic poses | 76 | 0.94 | 1.29 | 0.05 | 0.03 | 0.00 |
| Rowan-generated poses | 76 | 0.81 | 1.06 | 0.07 | 0.04 | 0.01 |
Conclusions
One of the big challenges in scaling FEP workflows, particularly in automated or semi-automated contexts like agentic science, is pose preparation. Traditionally, preparing poses for FEP is a difficult and labor-intensive process, involving significant manpower and limiting the extent to which FEP can be deployed in a black-box manner. We've been putting a lot of effort into building an automated pose-preparation pipeline, and we're starting to see results: we get good retrieval of experimental structures, and FEP runs done with Rowan-generated structures actually slightly outcompete experimental input structures.
Unfortunately for this blog post (but fortunately for the field), this benchmark set is challenging. While both FEP protocols generate good energy-based performance, the ability of RBFE to correctly rank the compounds in this set is relatively low. We expect that further protocol tuning or FEP improvements will be able to produce improved results; we haven't done any project-specific parameter tuning here and report these results simply as a zero-shot baseline about what's possible out of the box with FEP.
It's entirely possible that trivial modifications to our protocol will lead to improved performance. We've worked on this project for fewer than 24 hours, and report these data with the hope that others will do the same and push the field forward. Benchmarks drive scientific innovation, and tough and diverse benchmarks like OpenBind are exactly what's needed to push the free-energy field towards increased real-world predictive accuracy.
In the future, we plan to study which algorithmic improvements lead to better performance on this benchmark set, as well as working on automatic FEP protocol optimization, custom forcefield fitting per series, and studying additional benchmark systems from the OpenBind EV-A71 benchmark set. If you're interested in running FEP at scale, please reach out! We're always excited to build alongside new industry partners. Update: see our follow-up blog post here, where we discuss how to rescue performance on this dataset.
The complete data from this post are available on GitHub. If you're a free-energy-method developer, please consider testing this out and sharing your results! We're excited to continue learning about how we can do better on these challenging real-world datasets.