Predicting Ion-Mobility Mass Spectra Through Rowan
by Corin Wagen · Nov 5, 2025
Ion-mobility mass spectrometry is an advanced form of mass spectrometry (MS) that's often used in drug discovery and metabolomics. "Normal" MS works by ionizing the input sample, often via protonation, and then separating the resulting ions by their mass-to-charge ratio. Ion-mobility MS separations ions not only by their mass-to-charge ratio but also by how they move through a neutral gas under an electric field: compact ions drift faster, while extended ions bump into the gas more and drift more slowly. While different instruments implement this idea in different ways (drift tubes, traveling waves, and so on), the final output of ion-mobility MS is always a collision cross section (CCS), a calibrated value with units of Å2 that reflects how "big" the ion is in the gas phase.
Ion-mobility MS can be useful wherever assignment based solely on mass-to-charge ratio isn't enough. In metabolomics and lipidomics, CCS values can disambiguate structural and stereochemical isomers and improves library matching; in proteomics and native MS, ion mobility can report on protein and complex conformations and unfolding transitions. Because CCS is instrument-independent once calibrated, labs can build and share searchable libraries and use predicted CCS to filter candidates before chasing standards.
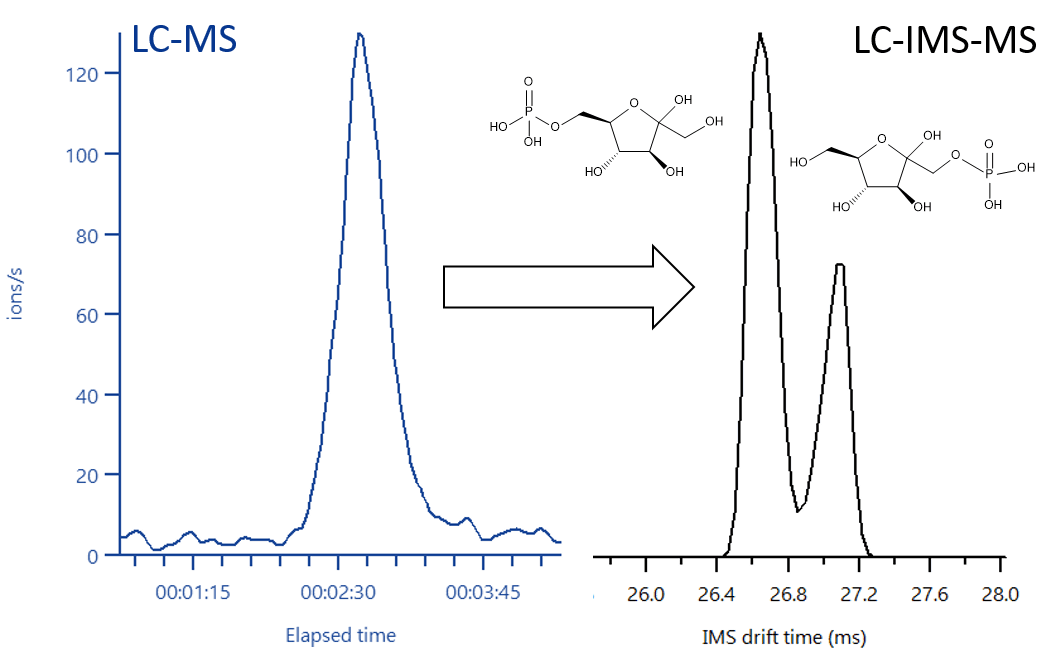
While these isomers can't be resolved by regular MS, they're easily resolved by ion mobility.
We became interested in the problem of CCS prediction over a year ago after talking to some industry users. While predicting out the mass-to-charge ratio for a given molecule is very simple and can be done in ChemDraw, figuring out what the CCS value for a given small molecule will be is quite tricky. If the molecule has already been reported, then its CCS value can be looked up in a database (like CCSBase): this works well for common molecules like amino acids, but obviously doesn't work for novel pharmaceuticals or agrochemicals.
If no experimental CCS data exists, then scientists can predict CCS values using machine learning or physics-based methods. There's been a lot of activity in using ML models for CCS prediction, but a recent benchmark from Sara de Cripan and co-workers argued that most ML-based CCS methods "suffer from a significant lack of generalization capacity" and are ill-suited for the type of metabolomics work that CCS prediction is often used for. Accordingly, we decided to focus on physics-based methods when implementing CCS prediction in Rowan.
Methods
Physics-Based CCS Prediction
The gold standard for physics-based CCS calculation is the "trajectory method," wherein the collision cross section is numerically simulated via thousands or millions of collisions with buffer gas molecules. This can be quite accurate but can take hours even for small molecules, even when using a fast forcefield to simulate gas–analyte interactions.
Running a trajectory-method calculation is just the final step in a successful CCS prediction— many more calculations have to happen to go all the way from structure to final CCS value. Here's how what running a single CCS prediction typically entails:
- The structure of the ionic adduct is determined. For [M+H]+ ions, this means identifying the site of protonation; for other ions, this is obviously different.
- A conformer search must then be run to identify all relevant gas-phase conformers. This conformer search needs to be quite careful, since CCS values are highly conformer-dependent (see this work from Das and Merz).
- The different conformers are optimized, scored, and assigned a Boltzmann weight. This is typically done with density-functional theory; for instance, MobCal MPI 2.0 uses ωB97X-D3/def2-TZVPP calculations.
- The CCS for each conformer is determined through trajectory-method calculations. As mentioned above, this can take hours per conformer!
- The final CCS value is computed through a Boltzmann-weighted average of each conformer's CCS value.
Rowan's Modifications
Ever since we started thinking about CCS prediction over a year ago, we've been brainstorming how we could speed this whole process up and make CCS prediction routine. Neural network potentials and semiempirical methods are much faster than density-functional theory, and so we thought that we might be able to accelerate the entire conformer optimization and screening step using NNPs or modern semiempirical methods. We conducted a variety of benchmark studies and found that, while AIMNet2 and other NNPs tended to overstabilize "closed" conformations of flexible gas-phase molecules and thus underestimate CCS values, the new g-xTB method from Stefan Grimme and co-workers was well-suited for scoring these conformers.
Unlike in previous Rowan workflows, though, just switching to low-cost computational methods for conformer optimization and screening wouldn't be enough to accelerate the whole workflow—we still needed to find a way to accelerate the slow trajectory-based CCS calculation. After some digging, we found CoSIMS, a simulation program that was able to compute helium-based CCS values significantly faster than MobCal (the standard trajectory-method implementation) by using a variety of tricks and clever approximations.

CoSIMS required a few modifications to fit within our workflow, though. Since most practical CCS measurements use nitrogen, not helium, we had to modify CoSIMS to allow the collision-gas parameters to be modified at runtime. Rowan's modified version of CoSIMS, available here, allows us to simulate nitrogen using a coarse-grained one-site model (similar to an approach investigated by Haack and co-workers).
We also developed a new forcefield by building hybrid scaled Lennard-Jones parameters for dinitrogen–small-molecule interactions derived from UFF, similar to the approach documented for MobCal-MPI using MMFF94 Lennard-Jones parameters. Specifically, we compute σ and ε for each dinitrogen–element pair by taking the geometric average of the per-atom σ and ε values from UFF and then applying a global scaling factor ρ. For dinitrogen, we approximate σ as 3.65 Å and ε as 0.29 kcal/mol.
Rowan's CCS Prediction Workflow
Rowan's final CCS-prediction workflow, which allows users to go directly from neutral SMILES strings to CCS values, works like this:
- Every protonation site on the molecule is automatically identified and protonated. This step can be skipped if the protomer is already specified.
- For each protomer, a careful conformer search is run using CREST, and every deduplicated output conformer is optimized using GFN2-xTB.
- The most stable protomer is identified by comparing the g-xTB energies of the optimized species. (If the protomer was manually specified, this step is also skipped.)
- Rowan's version of CoSIMS is used to compute the CCS value for each conformer with a predicted Boltzmann weight greater than 1%. AIMNet2-computed atom-centered charges are used as inputs for CoSIMS.
- The final CCS value is computed through a Boltzmann-weighted average of each conformer's CCS value.
This workflow generally takes a minute or two for small molecules, or up to half an hour for large and flexible molecules.
Benchmark Performance
In our hands, Rowan's CCS method generally gives absolute errors of about 5%. We ran our method against the [M+H]+ subset of the biomolecules reported by Xueyun Zheng and co-workers in their 2017 dataset and found that Rowan's CCS predictions gave a decent match with experiment, albeit with some systematic error. (If you're curious about where these errors come from and how they might be improved, check out our appendix below.)
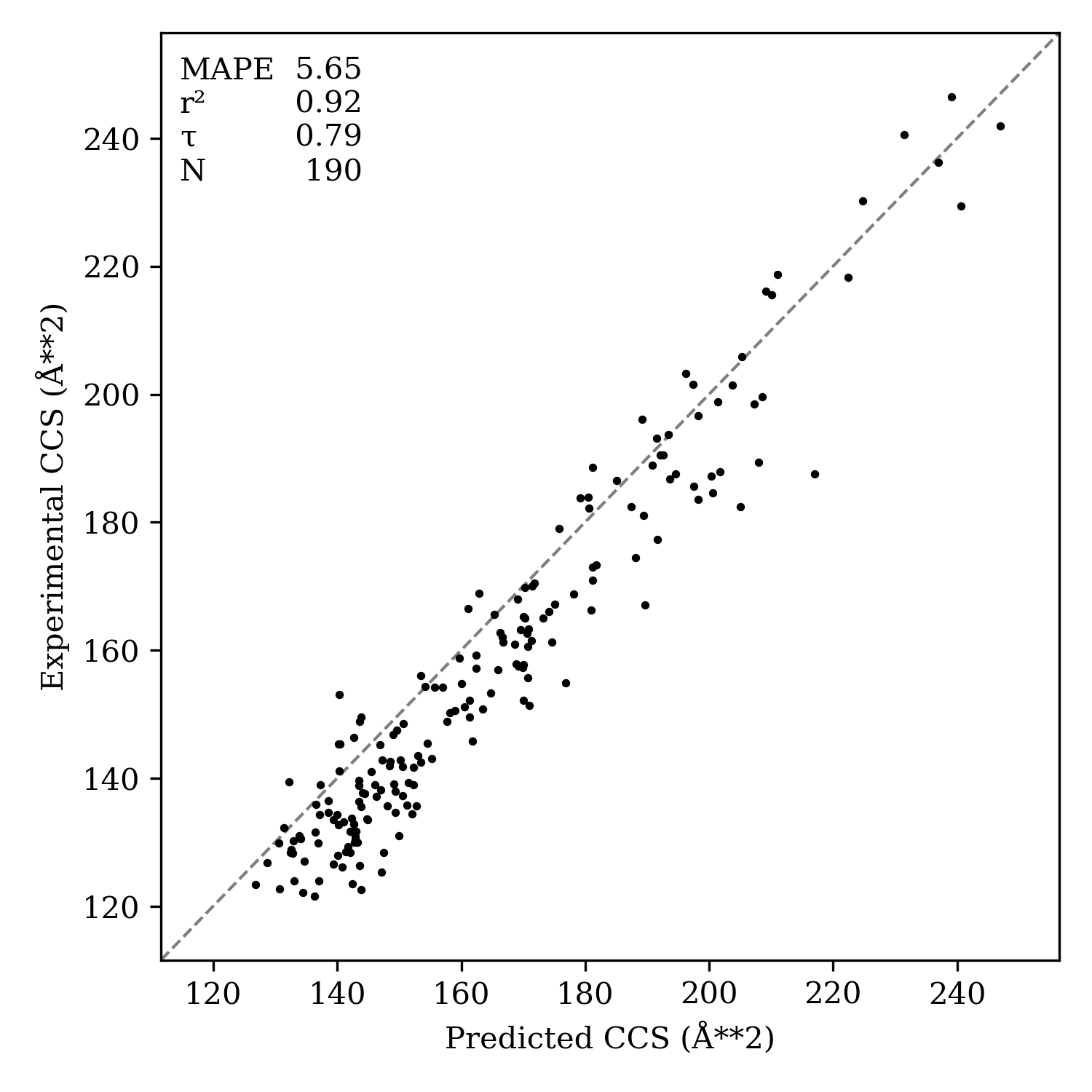
A comparison of Rowan's predicted CCS values to experimentally measured data.
Performance metrics on benchmarks is nice, but does Rowan's CCS workflow actually provide utility on real chemical problems? In a early pilot study with the Gair Group at Michigan State University, MSU researchers have found that Rowan's CCS predictions can be useful in quickly assigning isomeric mixtures that would otherwise require extensive isolation and characterization. Professor Joe Gair says:
Rowan has opened new research directions for our group. Comparing collisional cross sections calculated in Rowan versus those measured by ion-mobility mass spectrometry, we can assign structures to mixtures of diastereomers or regioisomers in an MS experiment that takes seconds.
While full details about how Professor Gair and co-workers are using Rowan's CCS predictions will have to wait until the publication of their paper, we're happy to be able to share some early real-world validation. This is just one use case—if you're interested in using Rowan's CCS predictions to accelerate your chemical analysis workflows, please reach out! We'd love to do a pilot study to understand the value that Rowan can bring to your scientific area.
Using Rowan's CCS Workflow
Subscribing Rowan users can run CCS predictions through the web-based GUI. Simply navigate to the ion-mobility mass spectrometry workflow, input the desired molecule, and click "Submit"—Rowan automatically allocates compute resources and runs the entire workflow described above, with no additional intervention needed.
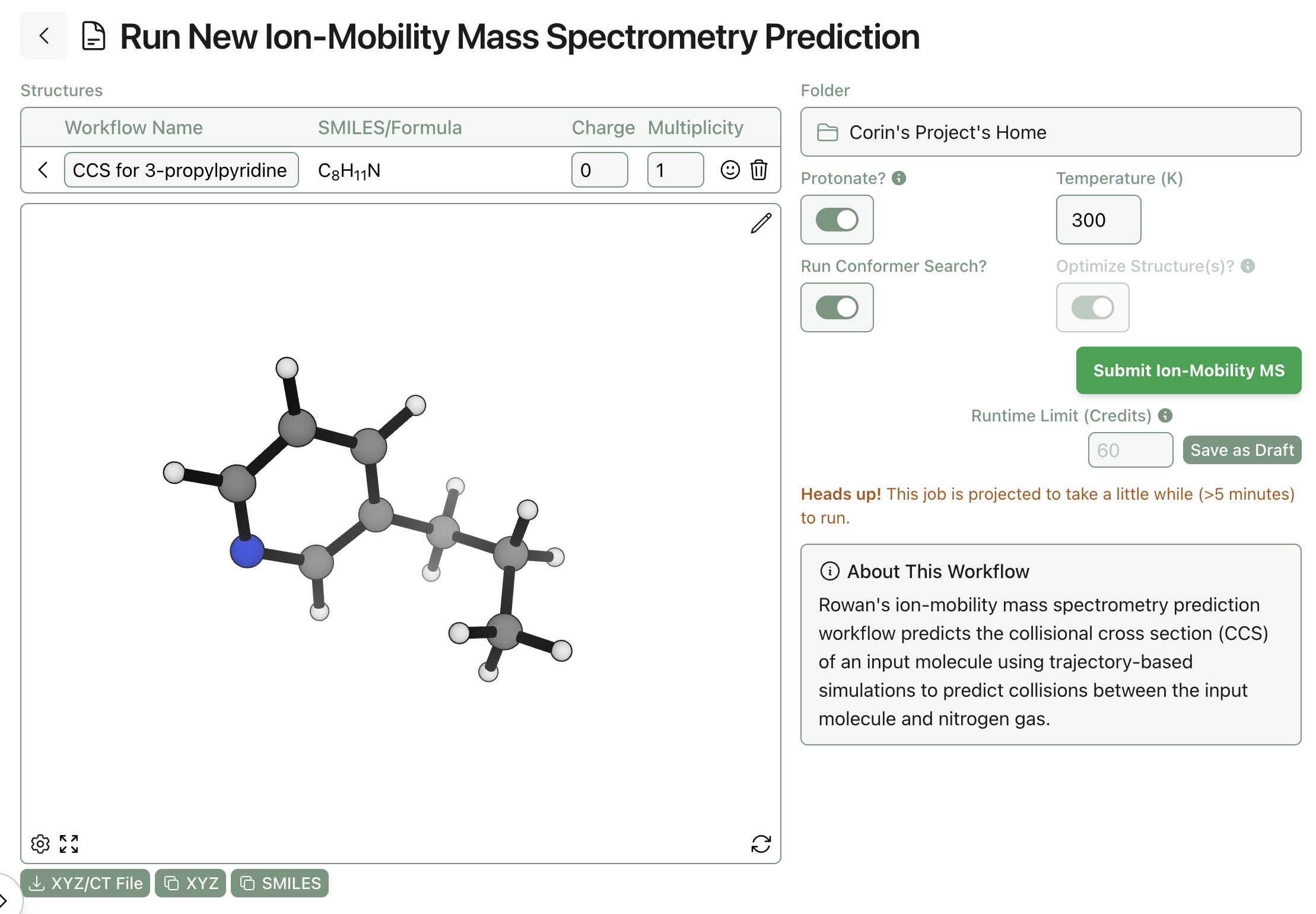
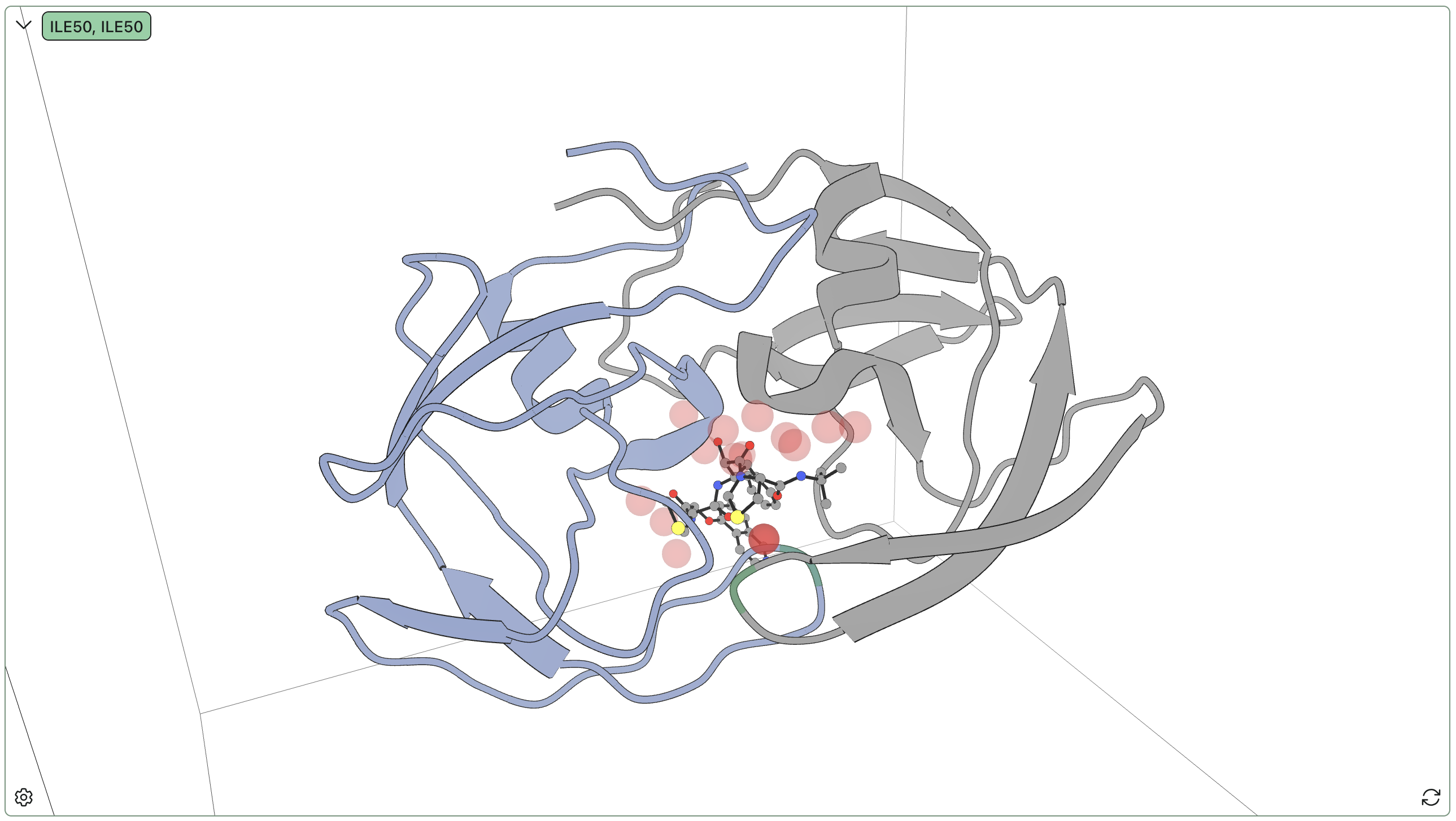
Submitting an ion-mobility MS prediction through Rowan.
The overall CCS prediction, alongside per-conformer predictions and weights, can be viewed on Rowan when the job is finished.
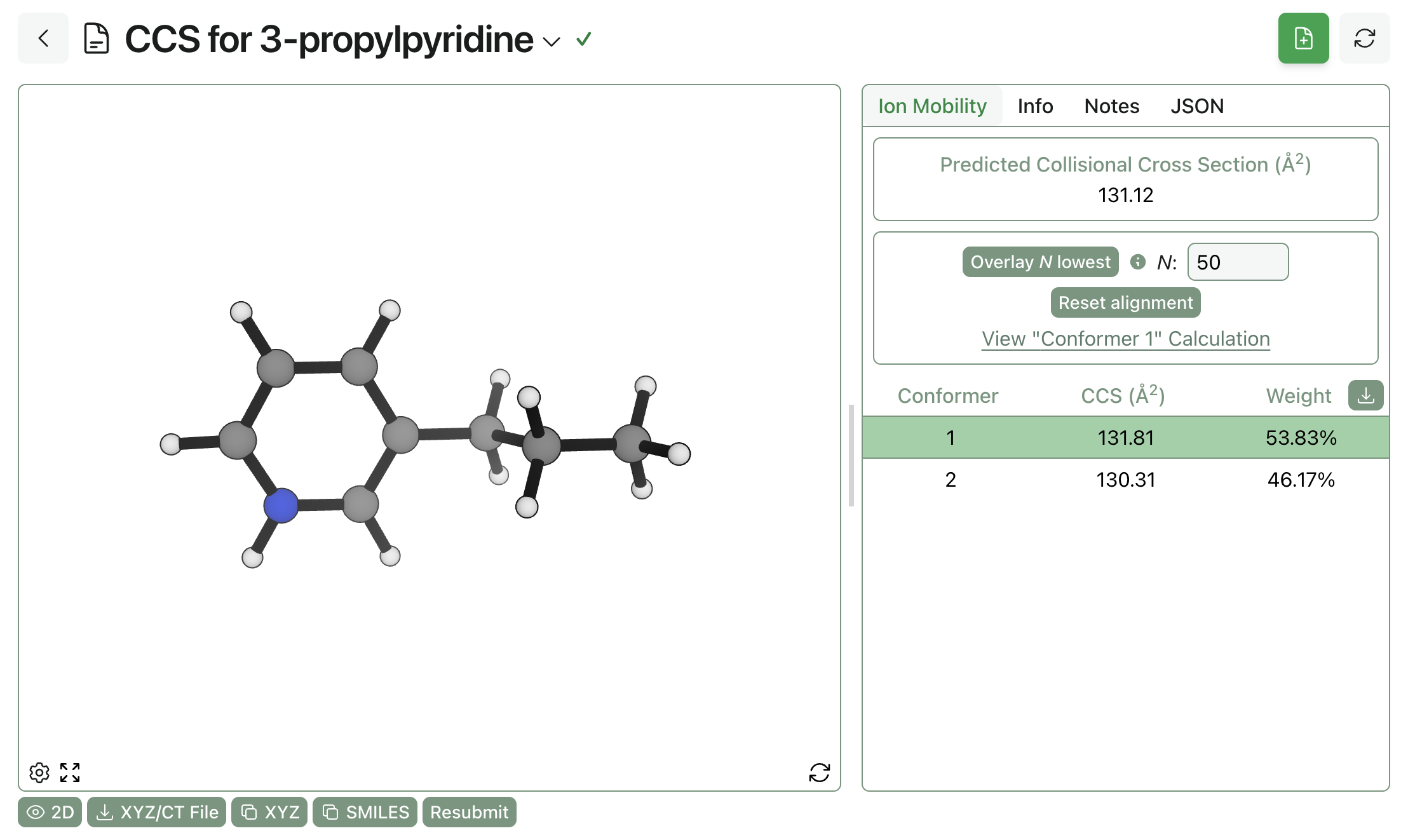
Viewing the result of an ion-mobility MS prediction through Rowan.
It's also easy to submit ion-mobility calculations through Rowan's API, allowing for high-throughput job submission and retrieval:
import rowan
import stjames
workflow = rowan.submit_ion_mobility_workflow(
stjames.Molecule.from_smiles("c1c(F)cccn1"),
protonate=True
)
workflow.wait_for_result().fetch_latest(in_place=True)
ccs = workflow.data["average_ccs"]
print(f"Predicted CCS: {ccs:.2f} Å**2")
This is just a simple example; a full explanation of how to tune various parameters through the API can be found in our documentation.
Appendix: Errors and Future Improvement
In the above section, we showed errors of about 5% in CCS prediction. Where do these errors come from?
One of the big sources of error, as highlighted by Das and Merz, is simply the completeness of the conformer ensemble. CCS values are acutely conformer-sensitive, and any inaccuracy in generating conformers (or scoring them) diminishes the accuracy of the output values. We've tried to strike a good balance between accuracy and speed, but generating complete conformer ensembles for large and flexible molecules remains a very difficult challenge and one that neither we nor the rest of the field have yet solved. As the accuracy of low-cost computational methods and conformer-generation methods improves (as we've written about elsewhere), we expect that Rowan's CCS workflow will naturally become more accurate.
Another challenge is forcefield construction. Many CCS forcefields are optimized for very small and rigid molecules where the complete conformer ensemble can be identified, like protonated aromatic heterocycles. In our hands, forcefields developed by fitting solely to these datasets work very well for small aromatic systems, but suffer from dramatic losses in accuracy when scaling to larger systems. (This may not be true for all approaches; we're just reporting what we've found.)
We've instead opted for a minimally parameterized UFF-derived forcefield, which is less accurate for small molecules but maintains higher generality for the large drug-like structures of interest to our users. This also lets us naturally incorporate new element types like boron and iodine, which are typically absent from CCS predictions. We recognize that this tradeoff is not optimal for everyone—users with a particular CCS forcefield that they've refit for their specific molecular data can submit custom forcefields via Rowan's API. If your company has a specific use case in mind and wants a custom fine-tuned CCS forcefield, please reach out!