Designing Protein and Peptide Binders with BoltzGen
Protein–ligand co-folding models like Boltz-2 tackle a "forward design" problem: given this collection of biomolecules, how will they bind to each other?
BoltzGen, a generative model developed by Hannes Stärk and co-workers from MIT (and other institutions), is trained to solve the corresponding "inverse design" problem: given a target and information about the binding mode, what combination of amino acids will bind? BoltzGen is built atop tools like Boltz-2 but additionally incorporates an all-atom diffusion model to generate new binder structures conditioned on a rich design specification language.
Distinctive Features of BoltzGen
Protein binder design is not a new field, and the success of BoltzGen has been enabled by many previous authors' work. Three years ago, work from David Baker and co-workers showed that miniprotein binders could be designed from a target protein structure, albeit with a low hit rate of about 0.5% (and worse in some cases). Subsequent work from Baker's group and others showed (1) that protein-folding methods like AlphaFold could be used to filter designs virtually, increasing hit rates substantially, and (2) that diffusion models could be used to generate backbones of protein binders, leading to dramatic efficiency improvements. Today, protein-binder-design models and workflows are routinely used in academia and industry with great success.
What differentiates the new BoltzGen model from existing protein-binder-design workflows like RFDiffusion, BindCraft, and Germinal?
Generality
Unlike other methods, BoltzGen uses a single model for designing proteins, peptides, and nanobodies against a variety of targets (including DNA, proteins, and small molecules). The authors argue that using one model for so many tasks will push the model to learn the correct underlying physics: "as models learn to emulate physics primarily through examples provided, we believe expanding the generality of the method further improves its design capabilities for specific classes as well" (from the paper).
While this generality seems simple, it's complex to implement: the BoltzGen model uses a 14-atom, geometry-based amino-acid representation for designed residues, making it possible to represent all structures at atomic resolution even before exact amino-acid residues have been determined. (The alternative is to combine discrete residue tokens with structure coordinates, which makes it complicated to jointly train models for both structure prediction and binder design.)
Tunability
BoltzGen employs a diverse design specification language which allows myriad aspects of the structure-generation process to be tuned: secondary structure, disordered regions, residue types, bonds, binding site, and so on and so forth. The authors see this tunability as a key advantage of their model:
Lastly, we comment on how there is a tendency in the field to claim that binder design models are "zero-shot" and "plug-and-play" solutions without a chance for failure. We do not make this claim and encourage users to use BoltzGen thoughtfully, carefully inspect the generated structures, and potentially rerun the pipeline multiple times, first at smaller and then larger scales. BoltzGen's rich design specification language provides a large degree of control that should be experimented with for optimal results.
(For more detail on how this design specification language works, see our accompanying piece on how to run BoltzGen.)
External Validation
BoltzGen's paper) includes extensive experimental validation from 26 external groups in academia and industry, including many challenging targets with high dissimilarity to training-data structures. These challenging prospective validation experiments show that BoltzGen can be used in difficult real-world contexts, unlike many in silico methods which struggle to generalize to the complexity of cutting-edge problems.
Applications of BoltzGen
The flexibility of BoltzGen makes the number of potential applications virtually limitless. Here's a few potentially relevant use cases:
- Protein-binder design. BoltzGen can be used to design miniprotein binders to specific sites on a target protein.
- Peptide-binder design. BoltzGen can also be used to design small peptides as binders to specific sites or regions on a protein, including cyclic peptides.
- Nanobody design. BoltzGen shows good success at designing single-domain antibodies, also known as nanobodies, towards challenging protein targets.
- Small-molecule-binding proteins. BoltzGen can also generate proteins that bind to arbitrary small molecules, which could plausibly be used for enzyme design or bioremediation.
How to Run BoltzGen
Rowan's web-based graphical user interface makes it easy to upload sequences, ligands, and structures, submit BoltzGen jobs to cutting-edge GPU hardware, and view & analyze the resulting predicted binders. The inputs and outputs of BoltzGen automatically integrate with the rest of Rowan's scientific workflows, including docking, co-folding, molecular dynamics, and protein preparation & sanitization, making it easy to run a state-of-the-art protein-design workflow without needing to build extensive scientific software infrastructure from scratch.
Here's what the output of a cyclic-peptide design run through BoltzGen looks like through Rowan:
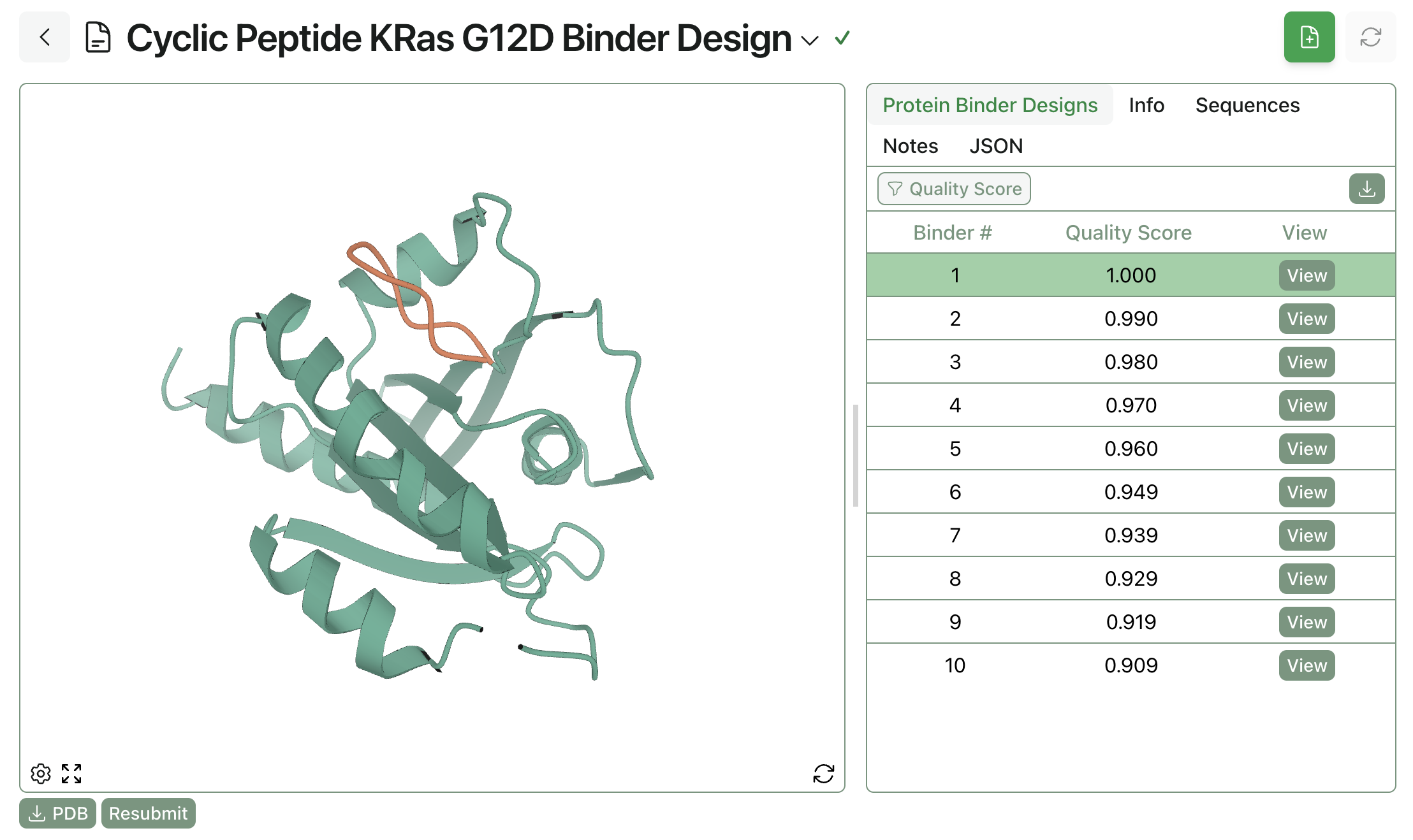
10 cyclic peptide binders to KRAS G12D, designed through Rowan.
Free-tier Rowan users can run up to 10 designs at once. If you're interested in running larger protein-design campaigns through Rowan's modern computational platform, please get in touch!
